

1 月 13 日,美國食品藥品監督管理局(Food and Drug Administration, FDA)發佈了一份指導草案,正式表態支持在藥物和生物製品臨牀試驗中使用貝葉斯方法論(Bayesian Methodology)。

圖 | 指導草案《在藥物和生物製品臨牀試驗中使用貝葉斯方法論》(來源:FDA)
所謂貝葉斯方法,是一種基於貝葉斯定理的統計推斷框架。其核心邏輯是:在觀察到新數據之前, 研究者對某個參數 (比如藥物在兒科人羣中的療效) 已有一定的先驗認知。
比如知道該藥物在成人羣體的藥效,這被稱爲“先驗分佈”:當新的試驗數據產生後,通過貝葉斯定理將先驗認知與觀測數據結合, 計算出更新後的“後驗分佈”,從而對參數做出概率性推斷。
簡單來說,貝葉斯方法允許研究者合法地、系統地利用歷史信息和外部證據,而不是每次試驗都從零開始。
FDA 局長馬蒂·馬卡里(Marty Makary)在聲明中直言:“貝葉斯方法論有助於解決藥物開發的兩大難題:高昂的成本和漫長的時間線。”這一政策調整源於《處方藥使用者付費法案》第七次重新授權(Prescription Drug User Fee Act VII, PDUFA VII)的承諾,旨在加速創新療法從實驗室走向患者牀邊。
圖 | 馬蒂·馬卡里發佈關於新指南的視頻(來源:X)
長期以來,FDA 臨牀試驗的設計與分析幾乎完全依賴頻率學派統計方法(Frequentist Statistics)。這套體系建立在假設檢驗和 p 值的基礎上——研究者需要預設一個“零假設”,然後通過試驗數據判斷能否以足夠小的概率(通常是 5%以下)推翻它。
頻率學派要求每個臨牀試驗都是相對獨立的統計單元,只能使用當前試驗中收集的數據進行推斷。歷史試驗的結果、真實世界的疾病數據、甚至已經發表的研究結論,都無法正式納入統計分析框架。
更關鍵的是,頻率學派給出的是一個二元判斷——要麼“有統計學顯著差異”,要麼“無顯著差異”——卻無法直接回答臨牀醫生和患者最關心的問題:這個藥物有多大概率真正有效?
尤其是在罕見病和兒科藥物開發中,頻率學派的限制格外突出。假設一種遺傳性疾病全球僅有數百名患者,按照傳統設計,一項具有統計效力的隨機對照試驗可能需要招募幾十甚至上百名患者才能達到“顯著性”標準,但這在現實中根本無法實現。即便勉強湊夠樣本量,試驗週期也會拖得極長。
以杜氏肌營養不良症(Duchenne Muscular Dystrophy, DMD)爲例,這種影響兒童的罕見肌肉疾病全球患者不足數萬人,且病情進展迅速。傳統頻率學派試驗設計要求數年隨訪才能觀察到足夠的臨牀事件,而患者的身體狀況根本無法等待。
此外,頻率學派嚴格的“試驗獨立性”原則意味着,即便某種療法在成人羣體中已經證明安全有效,但針對兒童的試驗仍需從零開始收集數據。這不僅浪費資源,也帶來倫理困境——明知可能有效的藥物,卻要讓一部分兒童接受安慰劑對照,或者因爲無法招募足夠受試者而讓整個兒科適應症開發計劃擱淺。
相較之下,貝葉斯方法提供了一種截然不同的思路。它的將“先驗信息”(Prior Information)與當前試驗數據結合,形成“後驗分佈”(Posterior Distribution),從而給出關於療效或安全性的概率陳述。
舉個具體例子:假設一款治療高血壓的藥物已經在 18 歲以上成人中完成了大規模臨牀試驗,證實能降低血壓且安全性良好。現在需要在 12-17 歲的青少年中進行試驗。
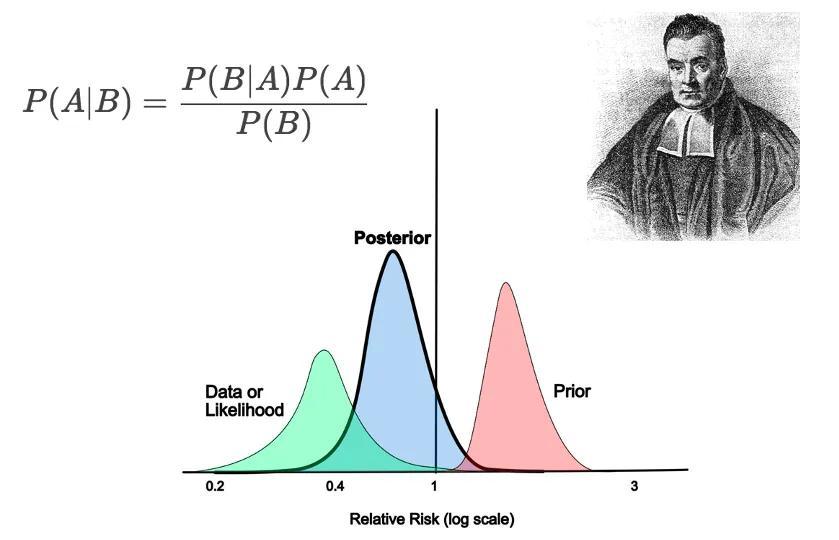
圖 | 貝葉斯統計示意圖及創始人托馬斯·貝葉斯(來源:Medium)
傳統頻率學派要求青少年試驗完全獨立設計,可能需要數百名受試者。而貝葉斯框架允許研究者將成人試驗的數據作爲先驗信息,基於藥代動力學和藥效學的相似性,合理推斷青少年羣體的預期療效範圍。這樣一來,青少年試驗只需要 50-100 名受試者就能獲得可靠結論,試驗時間也從 3 年縮短至 18 個月,同時推斷的精度反而更高,因爲整合了更多相關信息。
FDA 此次發佈的指導文件列舉了貝葉斯計算的幾種典型應用場景。第一,在適應性試驗(Adaptive Trials)中提前判斷無效性或成功。傳統試驗通常設定固定的中期分析時點,而貝葉斯框架可以根據累積數據動態計算“成功概率”和“無效概率”。如果某個治療組在招募 100 名患者後,後驗分佈顯示有效概率低於 10%,可以立即終止該組,將資源轉向更有希望的劑量或治療方案。
第二,爲後續試驗提供劑量選擇依據。早期探索性試驗往往樣本量有限,頻率學派分析可能無法給出明確的劑量建議。貝葉斯建模可以整合藥代動力學數據、生物標誌物反應和初步療效信號,計算出不同劑量在後續試驗中成功的概率分佈,幫助申辦方做出更有信息量的決策。
第三,整合外部對照組或非同期對照的數據。在某些罕見病中,由於疾病自然史數據充分且疾病進展模式穩定,可以使用歷史對照數據作爲先驗信息,減少甚至避免安慰劑對照組的設置。
第四,支持亞組分析,特別是在不同年齡、性別或基因型亞組中評估療效差異時,貝葉斯層次模型(Bayesian Hierarchical Model)可以“借用”總體數據的信息,提高小樣本亞組的估計精度。
第五,作爲試驗主要推斷的基礎,即用後驗概率陳述直接支持藥品上市申請。
這套方法最直接的受益者是罕見病和兒科藥物的開發團隊。FDA 在指導文件中特別強調,貝葉斯方法對於瞄準罕見病或兒科適應症的申辦方“尤其有價值”,因爲這些領域的患者羣體規模天然較小。
對於那些希望加速上市的創新療法,貝葉斯框架的靈活性意味着可以在試驗進行過程中動態調整設計。比如根據中期數據提前終止無效治療組,或者擴大有效劑量組的招募規模,從而縮短整體研發週期,降低成本。
研究估算顯示,傳統藥物從臨牀前研究到獲批上市平均耗時 10-15 年,總成本超過 20 億美元,其中臨牀試驗階段佔據了大部分時間和費用。如果貝葉斯方法能將 III 期試驗樣本量減少 30-40%,或將試驗時間縮短 6-12 個月,對整個行業的經濟效益和社會效益都將產生顯著影響。
但貝葉斯方法的應用,也伴隨着一系列挑戰和爭議。首先是“先驗信息”的選擇問題。如果先驗數據來自不同人羣、不同疾病階段或不同治療背景,直接借用可能引入偏倚,導致結論失真。比如成人與兒童的藥代動力學和藥效學特徵存在差異,兒童的代謝率通常更快,器官功能尚未完全成熟,某些藥物在成人中安全的劑量在兒童中可能產生毒性。
簡單外推成人數據作爲先驗,可能低估兒科用藥風險。因此,先驗分佈的構建需要深入的臨牀藥理學理解和嚴謹的統計建模,不能機械套用歷史數據。FDA 在指導文件中明確要求,申辦方必須充分論證先驗信息的“可交換性”(Exchangeability)——即歷史數據與當前試驗的相似程度,以及先驗權重的合理性。
其次,貝葉斯分析往往需要更復雜的統計建模和計算。儘管現代計算工具如 Stan、JAGS 等軟件已經大幅簡化了操作流程,但模型的構建、先驗分佈的設定、馬爾科夫鏈蒙特卡洛(Markov Chain Monte Carlo, MCMC)採樣的收斂診斷,以及後驗結果的解釋,仍然需要深厚的統計學和臨牀專業知識。
監管層面的擔憂同樣存在。頻率學派嚴格控制 I 型錯誤率(即錯誤地宣稱藥物有效的概率),通常設定爲 5%,這是藥品審批的重要安全閥。而貝葉斯方法在形式上不直接對應 I 型錯誤的概念,儘管可以通過模擬驗證其頻率學派操作特性(Frequentist Operating Characteristics),但監管機構仍需逐案評估是否滿足審批標準。
FDA 在指導文件中明確指出,申辦方在考慮貝葉斯設計時應儘早與監管部門溝通,必要時可通過“複雜創新試驗設計配對會議項目”(Complex Innovative Trial Design Paired Meeting Program)獲得額外的技術指導。這意味着,儘管政策大門已經打開,但具體能走多遠,仍取決於監管方與申辦方的共識程度和案例積累。
從全球視角看,歐洲藥品管理局(European Medicines Agency, EMA)也在同步推進類似議程。其 2025 至 2026 年工作計劃中明確提出要加速臨牀試驗,貝葉斯方法被視爲關鍵工具之一。
國際人用藥品註冊技術協調會(International Council for Harmonisation, ICH)早在 1998 年發佈的 E9 指南中就允許使用貝葉斯方法,前提是“理由充分且結果穩健”;而 2024 年定稿的 E11A 指南則進一步鼓勵在兒科藥物開發中應用貝葉斯外推。FDA 此次發佈指導草案,或者也是與國際監管趨勢對齊的戰略選擇。

(來源:ICH)
這份指導草案將在今年 3 月 13 日前接受公衆評論,隨後 FDA 將根據反饋修訂併發布最終版本。儘管 FDA 在 2010 年就發佈了針對醫療器械的貝葉斯指南,但在藥物開發中全面開放貝葉斯方法,仍需監管機構、學術界和產業界的持續磨合。對於那些手握創新靶點、卻苦於樣本量限制的研發團隊而言,這或許是一個值得抓住的窗口期。
參考鏈接:
1.https://www.fda.gov/news-events/press-announcements/fda-issues-guidance-modernizing-statistical-methods-clinical-trials?utm_medium=email&utm_source=govdelivery
3.https://www.nature.com/articles/s41573-023-00638-0
運營/排版:何晨龍



