

經過近 20 年的技術積累,RNA 測序領域迎來了一項重要突破。近日,美國密歇根大學區健輝(Kin Fai Au)教授團隊在 Nature Biotechnology 發表論文,提出了一種名爲 miniQuant 的新算法,有效解決了困擾科學界十多年的基因異構體(gene isoform)定量難題。
“這個問題大家已經討論了十幾年了,但沒有一個數學的、具有數據科學的定義。”區健輝向 Deeptech 表示。這一突破性工作不僅提供了嚴謹的理論基礎,還開發出實用的軟件工具,爲 RNA 測序數據分析帶來了新的標準。

圖丨區健輝(來源:區健輝)
在我們的認知中,一個基因往往對應一個蛋白質,但實際上,一個基因可以通過可變剪接(alternative splicing)產生多種被稱爲基因異構體的信使 RNA(mRNA)版本。這些異構體極大地豐富了生命的複雜性,但要準確地定量分析它們的表達水平,卻一直是生物信息學領域的一大挑戰。
目前主流的測序技術分爲二代和三代。二代測序如 Illumina 平臺讀長較短,通常爲 150 個鹼基對(base pair),但通量高、成本低;而三代測序如 PacBio 和 ONT(Oxford Nanopore Technologies)平臺讀長可達幾萬個鹼基對,能夠覆蓋完整的 RNA 分子,但通量相對較低、成本較高。因此,研究者們常常面臨一個兩難的選擇:究竟何時該用哪種技術?
這種技術選擇的困難源於一個根本問題:由於基因的不同異構體(isoform)之間共享外顯子序列,許多短讀段無法明確分配給其來源異構體,導致定量分析存在不確定性。而長讀段雖然能夠跨越完整轉錄本,但較低的測序深度又會影響低表達基因的檢測精度。
爲了科學地解決這一問題,研究團隊提出了 K 值(K-value)概念——一個基於廣義條件數(generalized condition number)的基因特異性指標,用於量化基因異構體定量過程中的不確定性。具體而言,K 值定義爲讀段類-異構體比對概率矩陣 A 的最大和最小正奇異值的比值。研究團隊通過嚴格的數學推導證明,在觀測誤差相對較小的情況下,相對定量誤差的上界與 K 值近似成正相關,K 值越高的基因越容易在異構體定量中出現誤差。
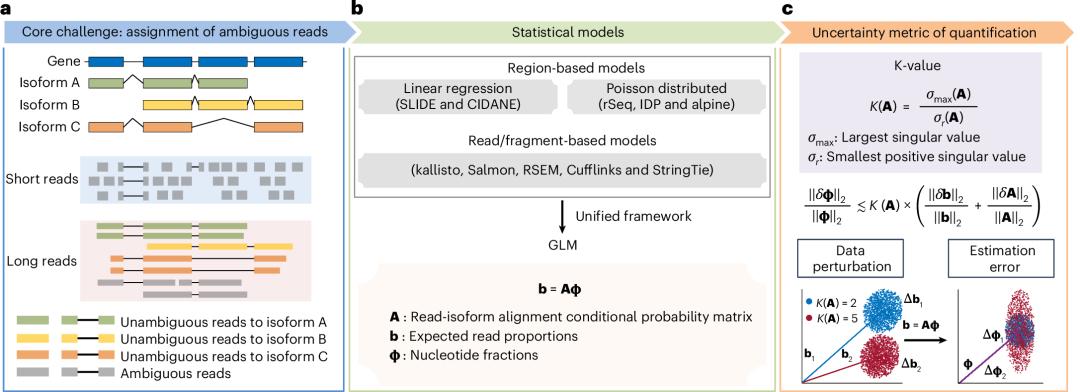
圖丨 K 值表示基因異構體定量中的不確定性(來源:Nature Biotechnology)
這一理論框架的重要性在於,它首次爲基因異構體複雜度的評估提供了數學上嚴格的定義。過去雖然有一些研究嘗試用異構體數量或外顯子數量來定義基因複雜度,但這些方法缺乏嚴謹的數據科學基礎。而 K 值的提出填補了這一理論空白,使得研究人員能夠在數據收集和分析之前就評估基因異構體定量的可靠性。
爲了驗證 K 值理論的有效性,研究團隊對超過 17,000 個來自 GTEx、TCGA 和 ENCODE 聯盟的公共數據集進行了大規模分析。結果顯示,當 K 值從 1 增加到 25 以上時,平均絕對相對差異(MARD,Mean Absolute Relative Difference)的中位數在 GTEx、TCGA 和 ENCODE 數據集中分別增加了 0.1830、0.1559 和 0.1721。更重要的是,這種關聯性在不同的生物學背景、樣本資源、測序平臺和數據質量條件下都保持一致,證明了 K 值作爲內在定量誤差指標的穩健性。
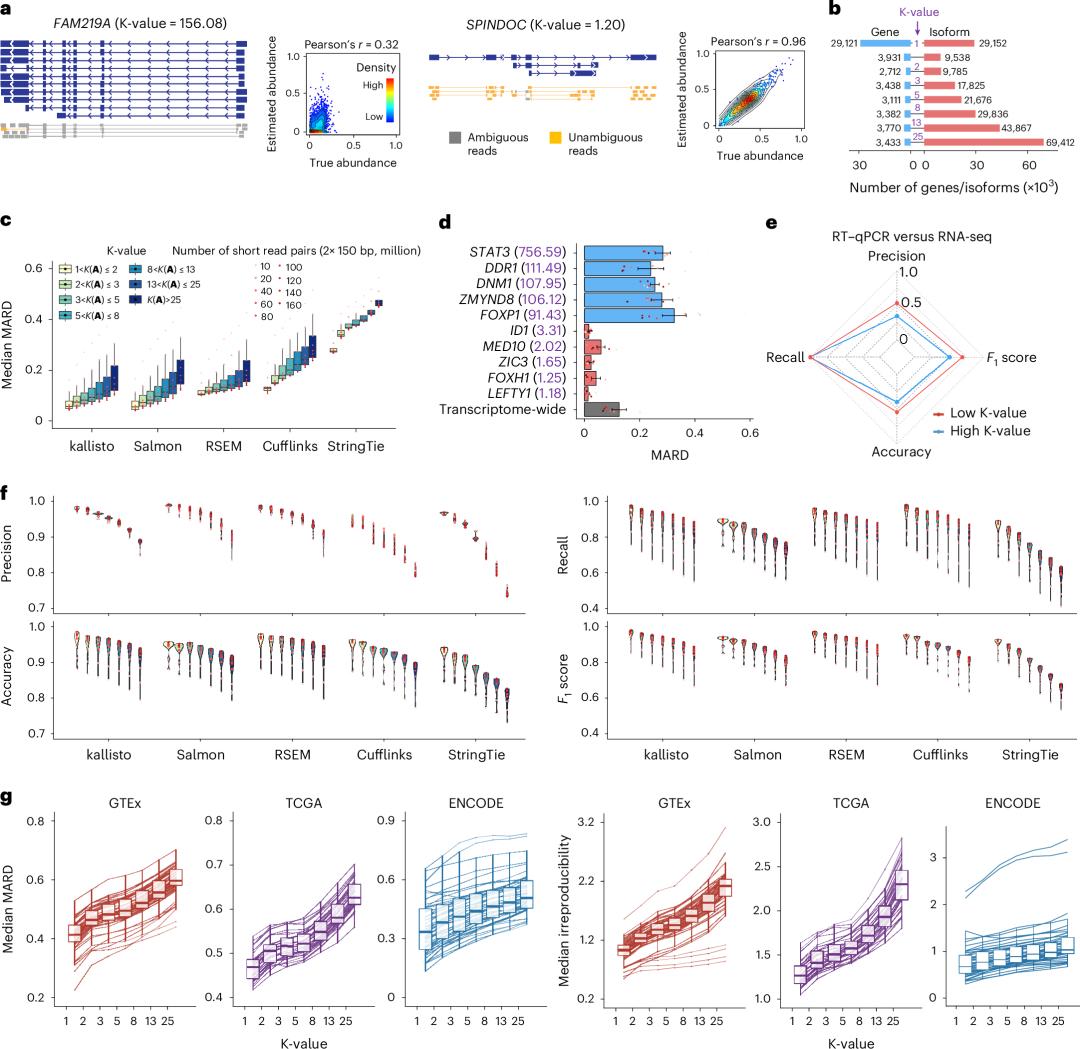
圖丨 K 值是基因異構體定量誤差的指標(來源:Nature Biotechnology)
基於 K 值理論,研究團隊開發了 miniQuant 軟件,這是一個能夠智能整合長短讀段優勢的創新工具。miniQuant 包含兩個模式:miniQuant-L 僅使用長讀段進行定量,而 miniQuant-H 則是核心創新的混合模式,能夠整合長短讀段數據。
區健輝介紹說:“我們可以用 K 值來幫助研究人員設計數據收集方案,指導他們應該選擇二代測序還是三代測序。”miniQuant-H 採用機器學習方法,根據基因結構特徵(包括 K 值)和數據特徵,爲每個基因羣體(gene community)確定最優的長短讀段權重組合。
該算法的智能之處在於,能夠針對不同的誤差來源採取相應的應對策略。對於結構複雜的基因(高 K 值),算法會更傾向於使用長讀段來減少解卷積誤差;而對於表達量較低的基因,則會增加短讀段的權重以減少採樣誤差造成的影響。例如,對於 K 值高達 82.26 且相對高表達的基因 VPS13D,miniQuant-H 分配了 0.75 的長讀段權重;而對於 K 值較低(5.37)且表達量相對較低的基因 TCP11L2,最優長讀段權重僅爲 0.25。
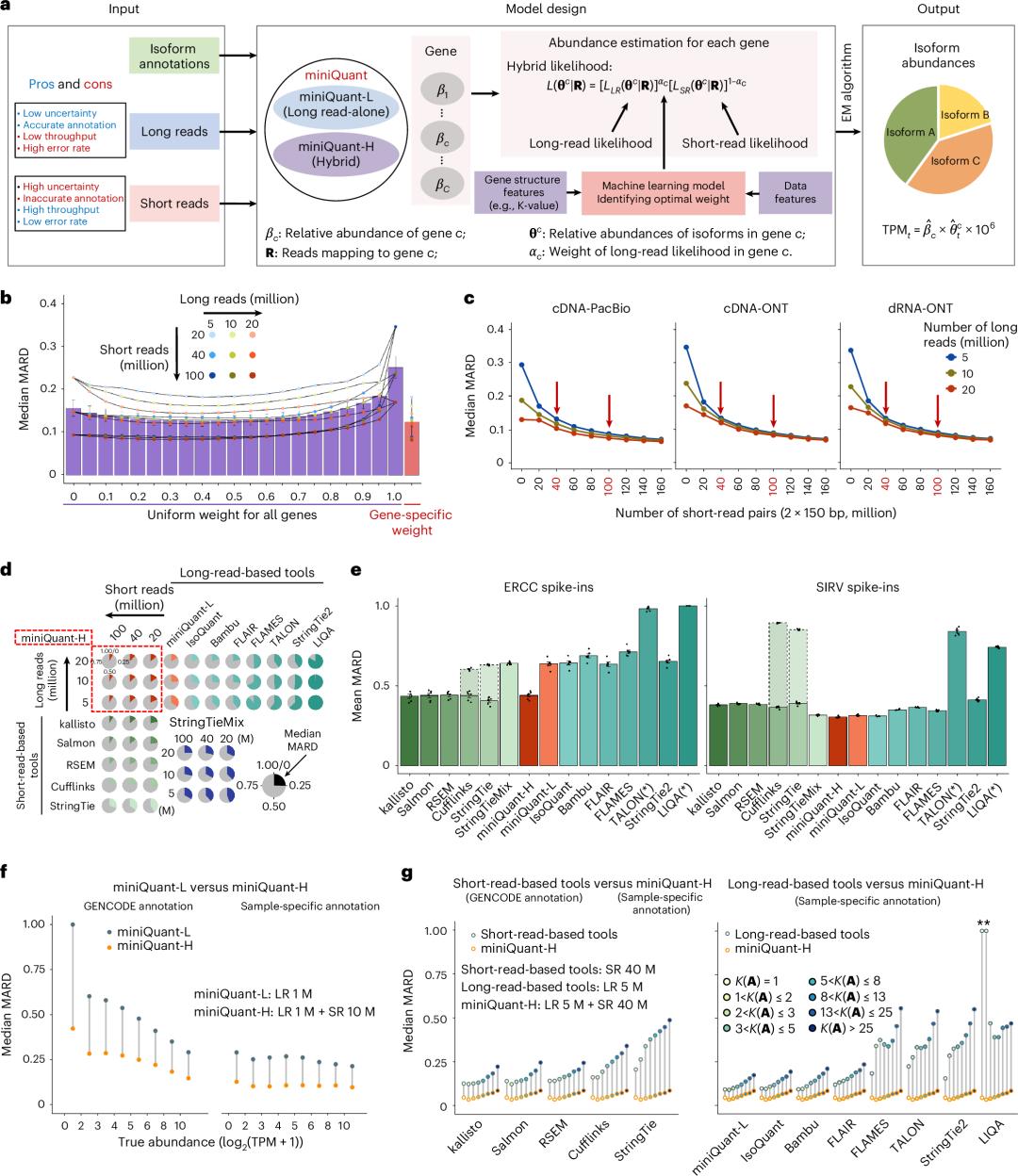
圖丨 MiniQuant-H 提升了基因異構體的準確定量(來源:Nature Biotechnology)
在多項基準測試中,miniQuant-H 展現出顯著優勢。在模擬數據測試中,該算法在多種測序深度組合下的平均中位數 MARD 爲 0.1249,顯著優於現有的短讀段工具(0.1505-0.3555)和長讀段工具(0.2515-0.9394)。在真實數據驗證方面,研究團隊使用了來自 LRGASP 聯盟的合成加標轉錄本數據。對於 ERCC 加標轉錄本,由於都是單異構體基因不存在解卷積誤差,長讀段工具由於採樣誤差普遍表現不如短讀段工具,而 miniQuant-H 達到了與短讀段工具相當的精度。對於結構複雜的 SIRV 加標轉錄本,長讀段工具表現更優,miniQuant-H 則取得了最低的平均誤差。
研究團隊進一步將 miniQuant 應用於人類胚胎幹細胞(ESC,Embryonic stem cell)分化研究中,揭示了幹細胞分化過程中的異構體轉換事件。通過分析 ESC 分化爲咽內胚層(PE,Pharyngeal Endoderm)和原始生殖細胞樣細胞(PGC,Primordial Germ Cell-like Cells)的過程,研究人員成功鑑定出 151 個(ESC 到 PE)和 161 個(ESC 到 PGC)發生異構體轉換的基因。這些發現具有重要的生物學意義,例如 MAT2B 基因雖然整體表達水平保持穩定,但其異構體使用模式發生了顯著變化,這種轉換可能影響細胞的凋亡調控能力。
值得注意的是,這些已識別的重要異構體轉換事件很多發生在高表達基因中(第 82-99 百分位,TPM 從 30.60 到 1,077.09),如果僅依靠長讀段測序,在典型的測序深度下(如 600 萬條 cDNA-ONT 讀段),當這些基因的表達水平被下采樣至第 75 百分位丰度時,長讀段工具的定量結果就會因爲採樣誤差而變得不可靠。相比之下,miniQuant-H 通過整合短讀段數據,能夠在更大的基因表達範圍內穩定地檢測異構體轉換模式。
與現有的整合方法相比,miniQuant 展現出明顯的技術優勢。例如,StringTieMix 採用相對簡單的讀段分配策略,將每個短讀段分配給最多長讀段支持的兼容異構體,在模擬數據中的表現有限。而 miniQuant-H 通過複雜的機器學習模型和聯合似然函數,實現了更精確和自適應的數據整合。
這項研究從兩個方面推動了 RNA 測序技術的發展:一方面,從理論上建立了評估基因異構體定量可靠性的數學框架;另一方面,在實踐上提供了能夠根據具體數據特徵和基因結構自適應選擇最優策略的軟件工具。
區健輝總結道:“這是首次以嚴謹的科學方法告訴研究人員哪些基因複雜、哪些基因簡單,以及何時應該選擇不同的測序技術。以前大家都是憑感覺和經驗來判斷,現在我們提供了科學的標準。”審稿人也評價該研究“回答了領域內長久以來懸而未決的問題”。
目前,miniQuant 軟件已在 GitHub 平臺(
https://github.com/Augroup/miniQuant)開源發佈,並提供了針對不同測序平臺和深度組合的預訓練模型,包括 cDNA-PacBio、cDNA-ONT 和 dRNA-ONT 等協議。隨着長讀段測序技術成本的持續下降和精度的不斷提升,這種智能整合長短讀段優勢的方法有望爲轉錄組研究提供更加精確和經濟的解決方案,推動基因異構體功能研究向更深層次發展。

圖丨相關論文(來源:Nature Biotechnology)
相關論文以《基於 miniQuant 的基因異構體定量優化方法》(Improving gene isoform quantification with miniQuant)爲題發表在 Nature Biotechnology 上 [1]。密歇根大學博士研究生李浩然、 王定傑、高琦、譚普文、王運浩和蔡曉羽博士是共同第一作者,區健輝教授擔任通訊作者。
參考資料:
1.Li, H., Wang, D., Gao, Q. et al. Improving gene isoform quantification with miniQuant. Nature Biotechnology(2025). https://doi.org/10.1038/s41587-025-02633-9
運營/排版:何晨龍



